
-
Mail us:
editor@raftpubs.org
Indexing & Abstracting
Full Text
Research ArticleDOI Number : 10.36811/jca.2020.110006Article Views : 16Article Downloads : 7
A Dft Pursuit on Tandem Cycloaddition-Cycloreversion of 2-Pyrone and 1,4-Oxazinone with Strained Alkynes
Akilandeswari L1, Kalpana P1 and Venuvanalingam P2*
1Department of Chemistry, Sri Sarada College for Women (Autonomous), Salem-636 016, Tamilnadu, India
2School of Chemistry, Bharathidasan University, Trichirappalli, Tamilnadu, India
*Corresponding Author: School of Chemistry, Bharathidasan University, Trichirappalli, Tamilnadu, India, Email: venuvanalingam@yahoo.com
Article Information
Aritcle Type: Research Article
Citation: Akilandeswari L, Kalpana P, Venuvanalingam P. 2020. A Dft Pursuit on Tandem Cycloaddition-Cycloreversion of 2-Pyrone and 1,4-Oxazinone with Strained Alkynes. J Chem Appl. 2: 10-20.
Copyright: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Copyright © 2020; Akilandeswari L
Publication history:
Received date: 29 July, 2020Accepted date: 27 August, 2020
Published date: 29 August, 2020
Abstract
2-Pyrone (2P) and 1,4-oxazinone (OXZ) react with strained alkynes viz., cyclohexyne (SA1), benzyne (SA2) and pyridyne (SA3) through concerted cycloaddition-cycloreversion reaction. All the reactions have been scanned and the activation parameters, FOE, deformation energies and variation in aromaticity computed at B3LYP/6-31g(d) level.The obtained results have been compared with that of acetylene. In SA1,faster cycloadditionis explained by its strain, in addition,aromatic gain is accountable to enhance the rate of cycloaddition in SA2 and SA3.
Introduction
Tandem cycloaddition-cycloreversion reactions are synthetically important to produce enormous natural products. One might reasonably expect that increased strain will lead to enhanced reactivity of a molecule and thus molecules become good candidates here to perform tandem reactions. Literature apparently shows the effect of ring size on the reactivity of cyclic alkynes [1-7]. Other strained alkynes (SAs) include arynes which are the reactive intermediates derived from substituted arenes that participate in some aromatic nucleophilic substitutions [8,9] and some cycloaddition reactions [10]. The most extensively studied aryne is benzyne which is known to undergo [4+2] cycloaddition with dienes like oxazole and thiophene derivatives [11-13]. Cyclohexyne has been reported to undergo tandem cycloaddition-cycloreversion with 2-pyrone to form tetrahydronaphthalene. This strategy when operated with some complex pyrones yielded analogues of planar frame work of dynemicin-A [14]. The effect of heteroatom on tandem cycloaddition-cycloreversion of 2-pyrone and 1,4-oxazinone with hetero alkynes has also been explored theoretically [15,16]. On the other hand,addition of benzynes and pyridynes with 2-pyrone have so far not been reported experimentally. This is the stimulus to have the aim of this work i.e., (i) to monitor the effect of strained triple bond of cyclohexyne and arynes over the tandem cycloaddition and cycloreversion, (ii) the effect of strain and aromatic gain on the reactivity in benzyne and pyridyne and (iii) the effect of strain, aromatic gain and the effect of hetero atom in pyridyne reaction. This study discusses the computed results such as activation parameters, FO analyses, deformation energies, variation in aromaticity along the reaction coordinates of the transition states formed during the tandem cycloaddition-cycloreversion reaction of 2P and OXZ with strained alkynes viz., cyclohexyne (SA1), benzyne (SA2) and pyridyne (SA3) and compared with that of acetylene.
Computational Methods
All the reactions have been modelled and scanned using Gaussian 98 program [17] at B3LYP [18,19]/6-31G (d) level. The magnetic susceptibility isotropy has been calculated by computing the NMR shielding tensors at B3LYP/6-311+G (2d, p) using IGAIM method [20,21] which is a slight variation of CSGT [20,21] method. Natural bond orbital (NBO) calculations have been performed. For both the cycloaddition and cycloreversion processes, the variation in magnetic susceptibility isotropy (χiso) along the reaction coordinate has been monitored. Change in aromaticity during the reactions has been computed.
Results and Discussion
Cycloaddition and cycloreversion of 2P and OXZ with strained alkynes SA1, SA2 and SA3 form benzo fused aromatics which are illustrated in Scheme 1 and 2. The relative free energy profile for the sequence of 2P and OXZ with strained alkynes are represented in figure 1-6 along with the optimized geometries and notable bond distances. There is the possibility of two extruding groups from the cycloadduct viz., CO2 and HCN in the case of cycloreversion of strained alkyne with OXZ. When SA3 is the dienophile there are 2 regioisomeric adducts possible, denoted as Regio 1 and Regio 2 in Scheme 1(c)& 2(c).
A. Energetics
2P-SAs reactions
The free energy profile in figure 2-5 and thermochemical parameters in table 4 indicate that 2P-SAs cycloadditions are much faster (barrier lying between 13-16 kcal mol-1) than 2P-Ace reaction (ΔG‡ for 2P-Ace cycloaddition is 36.84 kcal mol-1). Among strained alkynes considered, SA2 reacts faster (ΔG‡ =13.30 kcal mol-1) followed closely by SA3 (ΔG‡ =13.92 kcal mol-1) and then SA1which has relatively higher barrier by about 3 kcal mol-1 (ΔG‡ =16.70 kcal mol-1). In 2P-SA3 reaction, Regio 1 (R1)& Regio 2 (R2) pathways are closely lying with a very negligible difference (0.04 kcal mol-1). Hence the figure 4 shows the geometries of only R2 and relative free energies of the systems on R1 are given in parenthesis.
The cycloaddition of the three SAs lead to highly stable and exothermic cycloadducts viz., A1, A2 and R1A3 & R2A3. The exothermicity of these cycloadducts are (ΔHr= -65.55, -80.03, -77.27 kcal mol-1 of A1, A2 and R2A3 respectively) two to three fold greater than that of simple acetylene (ΔHr= -29.38 kcal mol-1) due to two factors: (i) Release of strain in the triple bond (ii) Aromaticity gain in the adduct. It is revealed by the formation of strainless aromatic benzene and pyridine fused with bicyclic lactones A2 and A3 respectively, in 2P-SA2 and 2P-SA3 reactions. In contrast, there is only strain release, not the aromatic gain in 2P-SA1 cycloaddition. The barrier to cycloreversion shown in table 1. indicates that A1 extrudes CO2 faster and is comparable with that of 2P-Ace reaction. On the other hand, A2 and A3 (ΔG‡ =15.95 kcal mol-1 and 16.91 kcal mol-1) have their cycloreversion barrier about 4-5 kcal mol-1 higher than 2P-SA1 reaction. This is ascertained to the excessive thermodynamic stability of the cycloadduct. Extra stabilization in A2 and A3 due to aromatic gain also results in higher barrier to cycloreversion.
OXZ-SAs reactions
The barrier to cycloaddition is reduced for OXZ-SAs reactions also, as observed in OXZAce. Reduction in barrier in OXZ-SAs reactions (ΔG‡=4.5 kcal mol-1, 3.3 kcal mol- 1and 2.8 kcal mol-1 for SA1, SA2 and SA3 respectively) are less than that of simple alkyne case (ΔG‡=7 kcal mol-1). Nitrogen in the system facilitates the reaction due to fewer aromatic loss and its remote location,from the reacting triple bond in SA3, affects the barrier to the least extent. Now the reactivity order of the SAs with OXZ is SA2>SA3>SA1>Ace. The reduction in free energy of activation of the OXZ-SAs reaction presented in table 2. It is ascribed to a much lesser activation enthalpy compared to 2P-Ace case shown in table 1. It is notable that ΔH‡ for the OXZ-SA2 and OXZSA3 reactions are slightly negative. ΔS‡ for cycloaddition and cycloreversion resemble that of 2P-Ace and 2P-SAs reactions. In OXZ-SAs reactionsSA2 and SA3 form cycloadduct A4 and A5, whereas SA3 makes two regioisomeric adducts R1A6 and R2A6. Their stability order is A4<R1A6-1 respectively) are similar to 2P-Ace reaction.
Frontier Orbital Analyses
Strained alkynes have their HOMO raised and LUMO lowered than simple alkyne and hence show higher reactivity. FOE gaps in figure 7 show that the 2P-SA1 cycloaddition follows Inverse Electron Demand (IED), similar to 2PAce. Added unsaturations and hetero atom stabilize the FOs of SA2 and SA3 respectively. As their LUMOs are lowered, SA2 and SA3 react by normal electron demand (NED) type. These FOE results are supported by QCT at the dienophile in the 2P-SA2 and 2P-SA3 reactions whereas 2P-SA1 reaction contradicts the FOE prediction, as its QCT is negative. In OXZ-SAs reactions, cycloaddition of OXZ-SA1 is IED type, similar to 2P-SA1 and is supported by QCT. Reduction in FOE gap indicates fall in the activation barrier to OXZ-SA1 cycloaddition. OXZ-SA2 reaction is IED, in contrast QCT predicts it to be a NED type as -0.028 charge is on the dienophile. FOE gaps show OXZ-SA2 reaction is neutral electron demand type whereas its QCT value is -0.047, representing it to be NED. In OXZ-SA3 and 2P-SA1 reactions, FOE gaps predicts IED type while QCT values indicate as NED type reactions.
Effect of strain on reactivity
The barrier to cycloaddition for 2P-SAs is less than 2P-Ace reaction, recognizing the reactivity (deformation energy) promoted by the strained triple bond. The deformation energies of diene and dienophile (DE1and DE2 respectively), in the corresponding transition state are presented in table 3. In 2P-Ace reaction, both diene and dienophile are largely deformed whereas drastically reduced in 2P with strained alkynes which has the order as SA1>SA3>SA2. This aligns their barrier trend.Further, in OXZ reaction N in the embedded diene still reduces the deformation energy of both diene and dienophile.
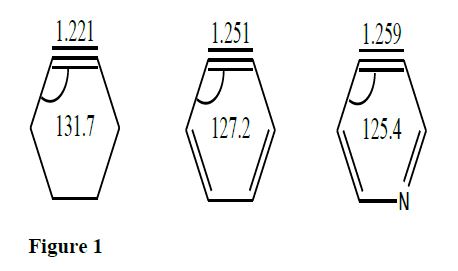
The bond length elongation and deviation of angle in SAs shown in figure 10 agree with the trend in the reactivity. The barrier to cycloaddition is also dependent on the difference between the initial angles (viz., C8-C7-C12 and C7-C8-C9) in the alkyne and that of cycloadduct. Comparison of variation of C8-C7-C12 and C7-C8-C9 angles for the strained and simple alkyne with 2P along the reaction coordinate of cycloaddition TS is presented in figure 11. Greater the difference in angle, greater is the barrier and variation in angle is sharp in 2P-Ace whereas very slight in 2P-SAs reactions.
Variation of aromaticity along the reaction coordinates
Aromatic gain during the reactions will definitely influence the rate. Among the three SAs involved reactions, SA2 and SA3 form benzo and pyrido-fused cycloadducts respectively, while SA1does not. The variation of aromaticity has been monitored by plotting the χiso along the reaction coordinate of both cycloaddition and cycloreversion of TSs. These plots of 2P-SA1 and 2P-SA2 reactions are collected in figures 14&15 respectively. Similar plots of 2P-SA3 and OXZ-SAs have not been done, as it resembles that of 2P-SA2.
2P-SA1 reaction
The change in χiso of TS1 (in the region -1 to +1 (amu)1/2 .Bohr) along the reaction coordinate is shown in figure 12(a). The curve indicates a continuous aromatic enhancement during the reaction, at the TS there is an increase (more negative χiso value representing aromatic enhancement) while the rate of aromaticity gain is gradually decreased. Figure 6 (b) & (c) show the variation of χiso of pyrone and cyclohexyne ring, indicating a decrease in negative χiso till TS followed by a steeper increase. It explains both the rings lose aromaticity during cycloaddition and hence the continual aromaticity increase in plot 6(a) is due to the pericyclic closed curve formed. In this case, gain in aromaticity even after the TS can be explained as follows. The reacting LUMO of SA1 is perfectly oriented for good overlap with incoming diene. Also the change in angles are less along the reaction coordinate as the SA1is nearly close to its geometry in the adduct. This is reflected in deformation energy (DE2) TS being very low for the SAs in general. Therefore, even after the TS, deformation of SA1 will be necessarily low thereby increase in the aromaticity. During cycloreversion, except for the absolute value of χiso which is very high due to the fused benzene, the trend remains the same. Both plots 6 (d) & (e) show a linear increase in χiso owing to the formation of tetrahydronaphthalene (P1) after CO2 extrusion and hence the aromaticity of newly formed ring dominates the scenario.
2P-SA2 reaction
The variation of χiso during 2P-SA2 reaction shown in figure 7 indicates a profile similar to 2P-SA1 reaction. In this case, a continual linear increase in χiso even after the TS is observed. It is ascribed to the aromatic gain by the benzene ring (plot 7 (c)). The pyrone ring fragment (plot 7 (b)) loses aromaticity which is compared with a low DE1 in 2P-SA2 reaction. Hence the least deformation of the pyrone ring is reflected in aromaticity monitored as variation in χiso. During cycloreversion, negative value of χiso increases due to CO2 extrusion to form naphthelene. Therefore, unlike in a typical pericyclic reaction, there is a continual increase of aromaticity.
|
Table 1: Thermochemical activation parameters of 2P-SAs reactions. (ΔG, ΔG‡, ΔH‡ and ΔHr are in kcal mol-1 and ΔS&ΔS‡ in cal K-1mol-1). |
||||||||||
|
Cycloaddition |
Cycloreversion |
|||||||||
|
DP |
Activation parameters |
Reaction parameters |
Activation parameters |
Reaction parameters |
||||||
|
ΔG‡ |
ΔH‡ |
ΔS‡ |
ΔHr |
ΔGr |
ΔG‡ |
ΔH‡ |
ΔS‡ |
ΔHr |
ΔGr |
|
|
SA1 |
16.7 |
4.48 |
-41.01 |
-65.55 |
-50.24 |
11.82 |
12.13 |
1.04 |
-50.67 |
-63.04 |
|
SA2 |
13.3 |
1.73 |
-38.83 |
-80.03 |
-65.04 |
15.95 |
16.42 |
1.58 |
-41.7 |
-54.13 |
|
SA3 |
||||||||||
|
(R1) |
13.96 |
1.99 |
-40.17 |
-77.02 |
-61.74 |
16.91 |
17.69 |
2.62 |
||
|
(R2) |
13.92 |
1.93 |
-40.24 |
-77.27 |
-61.98 |
15.84 |
16.34 |
1.68 |
-42.8 |
-54.99 |
|
Table 2: hermochemical activation parameters of OXZ-SAs reactions. (ΔG, ΔG‡, ΔH‡ and ΔHr are in kcal mol-1 and ΔS&ΔS‡ in cal K-1mol-1). |
|||||||||||||||
|
Cycloaddition |
Cycloreversion |
||||||||||||||
|
DP |
Activation parameters |
Reaction parameters |
CO2 Elimination |
HCN Elimination |
|||||||||||
|
ΔG‡ |
ΔH‡ |
ΔS‡ |
ΔHr |
ΔGr |
Activation parameters |
Reaction parameters |
Activation parameters |
Reaction parameters |
|||||||
|
|
|
|
|
|
|
ΔG‡ |
ΔH‡ |
ΔS‡ |
ΔHr |
ΔGr |
ΔG‡ |
ΔH‡ |
ΔS‡ |
ΔHr |
ΔGr |
|
SA1 |
12.14 |
0.17 |
-40.17 |
-60.71 |
-45.7 |
14.33 |
14.7 |
1.24 |
-47.3 |
-59.7 |
29.25 |
30.43 |
3.96 |
-0.32 |
-13.35 |
|
SA2 |
10.27 |
-0.87 |
-37.38 |
-88.69 |
-73.61 |
19.69 |
20.23 |
1.81 |
-38.34 |
-50.83 |
39.17 |
40.64 |
4.9 |
15.96 |
2.69 |
|
SA3 |
|||||||||||||||
|
(R1) |
11.29 |
-0.34 |
-39.03 |
-84.89 |
-69.52 |
19.37 |
19.95 |
2.05 |
-39.3 |
-69.34 |
39.24 |
40.71 |
4.32 |
-14.42 |
1.09 |
|
(R2) |
11.16 |
-0.54 |
-39.26 |
-85.29 |
-69.93 |
21.34 |
21.89 |
1.85 |
-38.28 |
-51.78 |
39.58 |
40.98 |
4.07 |
-17.58 |
4.28 |
|
Table 3: Deformation energy of dienes (2P & OXZ) (DE1) and dienophile (Ace, SA1, SA2, SA3) (DE2) components in the concerted cycloaddition transition states in (kcal mol-1) and quantum of charge transferred to dienophile |
||||
|
Diene |
DE1 |
Dienophile |
DE2 |
QCT |
|
|
19.79 |
Ace |
11.74 |
0.024 |
|
2P |
7.80 |
SA1 |
0.77 |
-0.001 |
|
|
4.37 |
SA2 |
0.60 |
-0.069 |
|
|
4.71 |
SA3 |
0.67 |
-0.090 |
|
|
16.86 |
Ace |
9.33 |
0.052 |
|
OXZ |
4.65 |
SA1 |
0.39 |
0.029 |
|
|
1.90 |
SA2 |
0.23 |
-0.028 |
|
|
2.33 |
SA3 |
0.27 |
-0.047 |
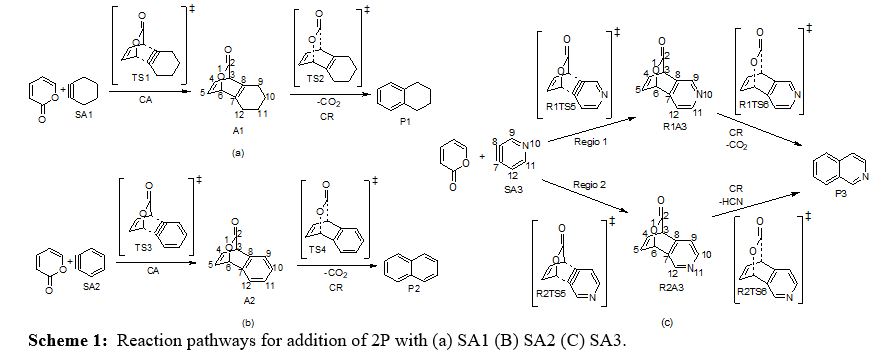
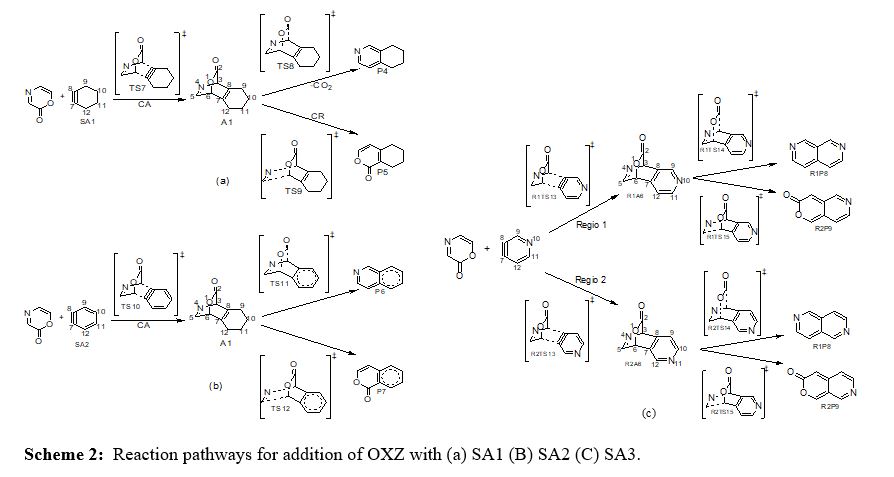
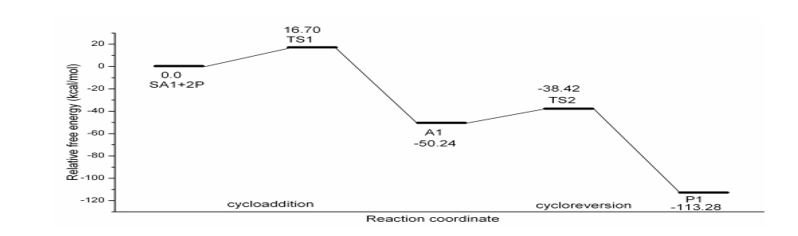
Figure 2: Schematic free energy profile of 2P-SA1 reaction computed at B3LYP/6-31G (d) level.
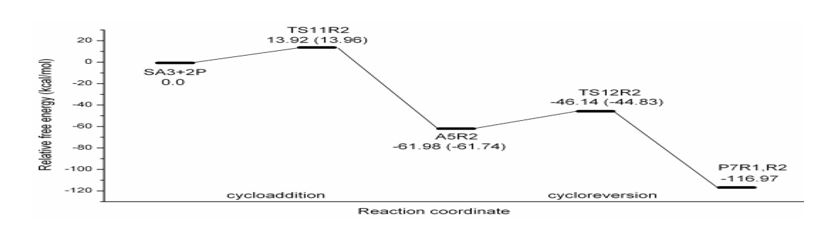
Figure 3: Schematic free energy profile of 2P-SA3 reaction computed at B3LYP/6-31G (d) level (The values in the brackets refer to Regio 1 reaction).
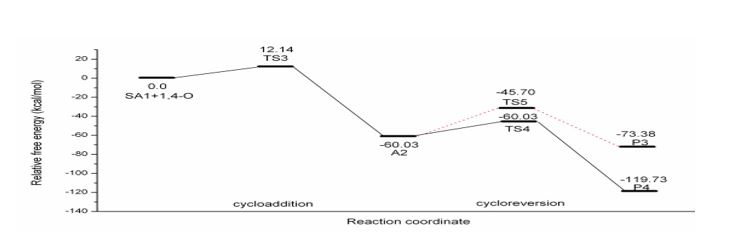
Figure 4: Schematic free energy profile of OXZ-SA1 reaction computed at B3LYP/6-31G (d) level.
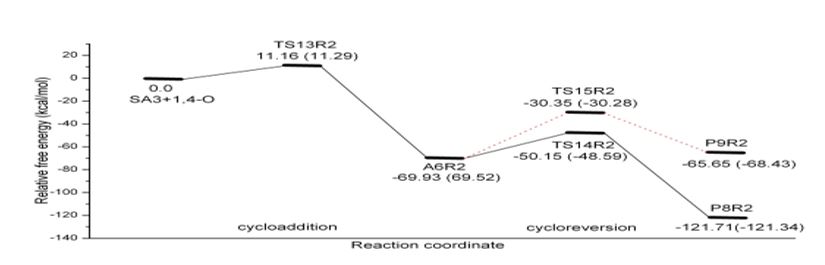
Figure 5: Schematic free energy profile of OXZ-SA3 reaction computed at B3LYP/6-31G (d) level (The values in the brackets refer to Regio 1 reaction).
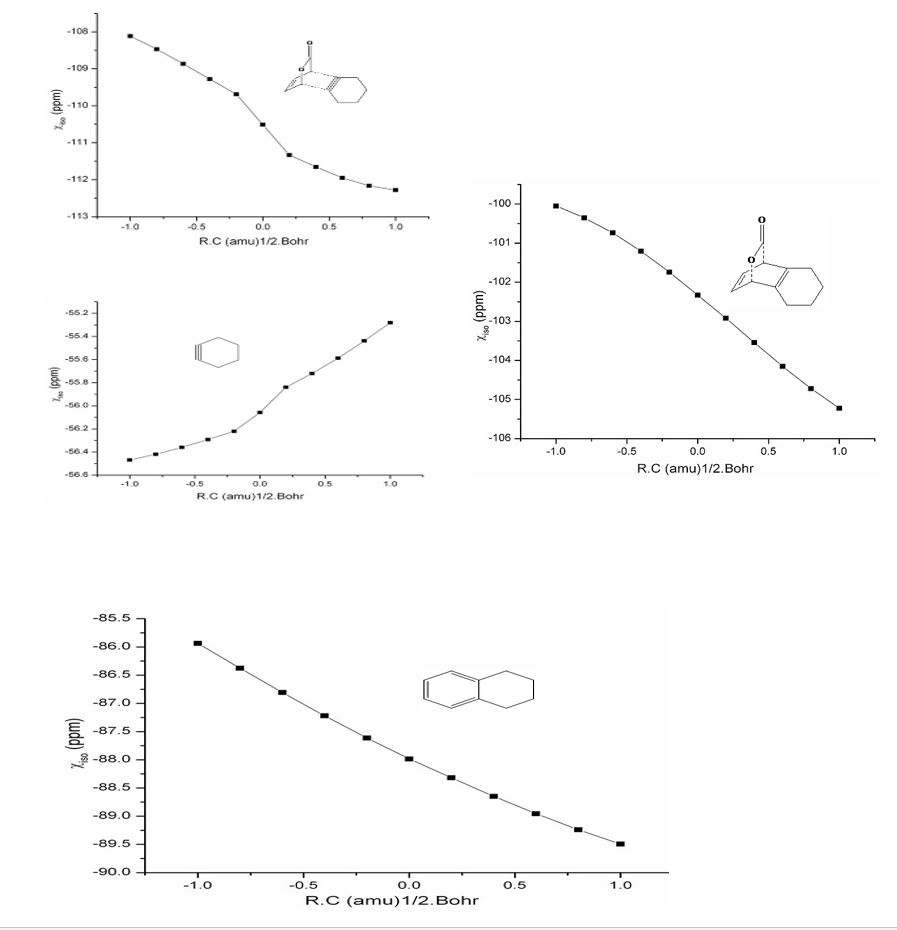
Figure 6: Variation of magnetic susceptibility isotropy along the reaction coordinate of 2P-SA1reaction around (a) TS1 (b) pyrone ring of TS1 (c) cyclohexyl ring of TS1 (d) TS2 (e) tetrahydronaphthalene ring of TS2.
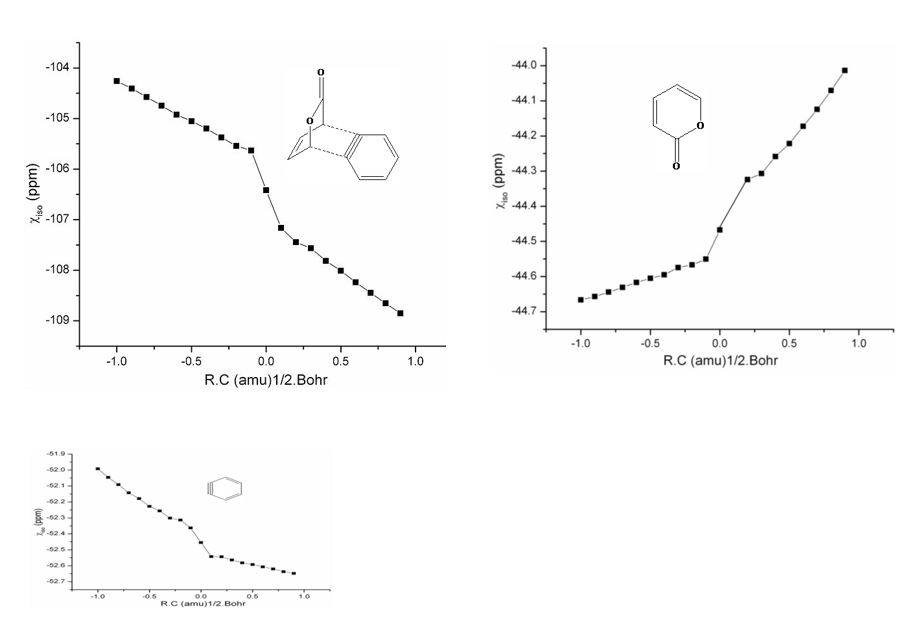
Figure 7: Variation of magnetic susceptibility isotropy along the reaction coordinate of 2P-SA1reaction around (a) TS1 (b) pyrone ring of TS1 (c) benzene ring of TS1 (d) TS2 (e) naphthalene ring of TS2.
Conclusions
Tandem cycloaddition-cycloreversion of 2-pyrone and 1,4-oxazinone with strained alkynes namely cyclohexyne(SA1), benzyne(SA2) and pyridine(SA3) occurs through concerted mechanism with slight asynchronicity due to the unsymmetric diene. Among the strained alkynes, benzyne reacts faster and pyridyne follows it, while cyclohexyne does slowly. All the three strained alkynes react extremely faster than Acetylene (Ace). Cycloreversion of 2P-SA1 adduct is comparable to 2P-Ace. In SA2 & SA3, cycloreversion is the rate determining step owing to the larger stability of adducts by strain release and aromatic gain by small ΔH‡ and magnetic anisotropy variation. Aza diene cycloadds faster with strained alkynes whereas cycloreverts slower than the non aza diene. Deformation analysis explains extremely faster cycloaddition of SA1 with 2P & OXZ is due to its strain. In SA2 & SA3, strain and aromatic gain enhance the rate of cycloaddition and is demonstrated by variation of aromaticity. FO analysis shows the normal and inverse electron demands in these dienophiles especially in benzyne and pyridyne. Overall, the study proves that release of strain and aromaticity gain governs the tandem reaction of strained alkynes.
References
1. Wittig G. 1942. Phenyl-lithium, der Schlüssel zu einer neuen Chemie metallorganischer Verbindungen Naturw is sensch aften. 30: 696. Ref.: shorturl.at/jCDKY
2. a) Hoffmann RW. Dehydrobenzene, Cycloalkynes. 1967 Academic Press: New York. b) Krebs A. 1969. In Chemistry of Acetylenes, Marcel Dekker: New York. c) LeNoble WJ. 1974. Highlights of Organic Chemistry; Marcel Dekker: New York. d) Greenberg A, Liebmann JF. 1978. Strained Organic Compounds; Academic Press: New York. e) Wittig G. 1963. Pure Appl. Chem. 7: 173.
3. a) Nakagawa M. 1978. in The Chemistry of the Carbon-Carbon Triple Bond. Wiley: New York. b) Montgomery, L, Roberts JDJ. 1960. Am. Chem. Soc. 82: 4750, Wittig G, Wenlich J, Wilson E. Chem. Ber. 1965. 98: 458. Gassman, P.; Gennick, I. J. Am. Chem. Soc. 1980, 102, 6863.
4. Montgomery LK, Applegat LEJ. 1967. Am. Chem. Soc. 89: 5305.
5. Allinger N L, Meyer AY. 1975. Conformational analysis-CIX: Applications of molecular mechanics to the structures and energies of alkynes. Tetrahedron. 31: 1807. Ref.: shorturl.at/cprMW
6. Schmidt H, Schweig A. Krebs A. 1974. Splitting of the degenerate acetylenic πmos; a probe for ring strain. Tetrahedron Lett. 16: 1471. Ref.: shorturl.at/ftGS6
7. a) Bunnett JFJ. 1961. Chem. Educ. 38: 278. b) Wittig, G. 1961 Angew. Chem. Int. Ed. Engl. 4: 731.
8. Gilchrist TL, 1983. in The Chemistry of functional groups, Supplement C. Wiley: New York.
9. Bryce MR, Vernon JM. 1981. Reactions of Benzyne with Heterocyclic Compounds. Adu. Heterocycl. Chem. 28: 183. Ref.: shorturl.at/htQWX
10. Dolbier WR, Loma D, Garza T, et al. 1972. Dimerizations of methylenecyclopropanes Harmon. Tetrahedron. 28: 3185. Ref.: shorturl.at/glIJP
11. van der Plaas HC, Roeterdink F. 1983. in The Chemistry of functional groups, Supplement C. Wiley: New York.
12. Whitney SE, Winters M, Rickborn BJ. 1990. Org. Chem. 55: 929.
13. Thiemann T, Fujii H, Ohira D, et al. Cycloaddition of thiophene S-oxides to allenes, alkynes and to benzyne. 2003. New J.Chem. 27: 1377. Ref.: shorturl.at/csHV3
14. Atanes N, Escudero S, P?rez D, et al. Generation of cyclohexyne and its Diels-Alder reaction with α-pyrones. 1998. Tetrahedron Lett. 39: 3039. Ref.: shorturl.at/kqrBE
15. Akilandeswari L. Ph.D thesis. 2007. Bharathidasan University Tiruchirappalli,https://shodhganga.inflibnet.ac.in/jspui/handle/10603/115127.
16. Akilandeswari L, Kalpana P. 2018. International Journal of Chemistry Studies. 2: 12.
17. Frisch MJ, et al. 1998. Gaussian 98 (Revision A.9), Gaussian, Inc. Pittsburgh, PA.
18. Becke AD. 1993. Density?functional thermochemistry. I. The effect of the exchange?only gradient correction. Chem. Phys. 98: 5648. Ref.: shorturl.at/cnCR0
19. Lee C, Yang W, Parr RG. 1988. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 37: 785. Ref.: shorturl.at/bsLQ7
20. Keith TA, Bader RFW. 1993. Chem. Phys. Lett. 220: 223.
21. Keith TA, Bader RFW. 1992. Chem. Phys. Lett. 194: 1. Ref.: shorturl.at/eGHM5
22. Cheeseman JR, Frisch MJ, Trucks GW, et al. 1996. Chem. Phys. 104: 5497. Ref.: shorturl.at/aBX05


















