
-
Mail us:
editor@raftpubs.org
Indexing & Abstracting
Full Text
Case ReportDOI Number : 10.36811/ijgmgt.2019.110003Article Views : 13Article Downloads : 15
Genetic counselling of Mucopolysaccharidosis type III: case report and literature review
Jamileh Malbin1,2*, Shahab Galeban2, Yosef shafagati5, Mohammad sadegh fallah3 and Sirous Zeinali3,4
1Azerbaijan National Academy of Science (ANAS), Genetic Resources Institute, Baku, Azerbaijan
2Genetic counseling center, Urmia Welfare Organization of West Azerbaijan Iran
3Kawsar Human Genetics Research Center (KHGRC), Tehran, Iran
4Department of Molecular Medicine, Biotech Research Center, Pasteur Institute of Iran, Tehran, Iran
5Cell Research Center, Sarem Women Hospital, Tehran Iran
*Corresponding author: Jamileh Malbin, Genetic Counseling Center of Welfare Organization of West Azerbaijan, Iran; Email: jamaalbin@yahoo.com
Article Information
Aritcle Type: Case Report
Citation: Jamileh Malbin, Shahab Galeban, Yousef Shafeghati, et al. 2019. Genetic counselling of Mucopolysaccharidosis type III: case report and literature review. Int J Genet Med Gene Ther. 1: 09-14.
Copyright: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Copyright © 2019; Jamileh Malbin
Publication history:
Received date: 23 May, 2019Accepted date: 15 June, 2019
Published date: 16 June, 2019
Abstract
Mucopolysaccharidosis type III (MPSIII), or Sanfilippo syndrome is a lysosomal storage disease with neurological manifestations in affected children. It is heterogeneous diseases in all aspect; prevalence, clinical presentation, biochemistry and causative mutations. There are four biochemical forms of this disease: MPSIIIA, B, C, and D which each one has a different gene and involving enzyme. Molecular genetics and enzyme activity assay has been used to diagnosis of MPS and recently next generation sequencing technique has been used by some researcher. Here we report the family with two affected child which has been diagnosis by enzyme function analysis and counseling challenges for prenatal diagnosis in this family.
Keywords: Mucopolysaccharidosis; Enzyme activity test; Prenatal diagnosis
Introduction
Lysosomal storage disorders (LSDs) are rare inherited debilitating and often fatal disorders caused by mutations affecting lysosomal proteins and function. [1]. Mucopolysaccharidoses (MPSs) are a group of rare lysosomal storage disorders caused by deficiency of enzymes catalyzing the stepwise degradation of glycosaminoglycans dermatan sulfate, heparan sulfate, keratan sulfate, chondroitin sulfate, and hyaluronic acid [1,2]. MPS III or Sanflippo syndrome is an autosomal recessive disease [1,3] with four different subtypes (A, B, C, and D) caused by mutations in different proteins (table 1) [1,4]. Patients are asymptomatic at birth but develop neurological problems in early childhood. In particular, behavioral abnormalities are followed by intellectual deterioration and dementia, and life expectancy is severely reduced. There are a few treatment option for some kind of LSD but it is very expensive so for the couple with affected child prenatal diagnosis still is the first choice [5].
Many methods have been used to approach a diagnosis in the MPSs. Laboratory assays based on quantification of the accumulated products is one of the most favored methods [6]. Enzyme activity assay in dried blood spots could be very useful in the diagnosis of a MPS but usually needs verification with a second type of assay [7] but are usually not reliable in detecting heterozygous carriers. While biochemical methods are still the gold standard in pre and postnatal diagnosis, they are often very difficult and time consuming; besides it requires precise clinical diagnosis to reduce the number of biochemical tests for each patient [2,6,7]. Even having achieved a diagnosis with biochemical assays, establishing a molecular genetic diagnosis is still important step especially for those which new therapies could be available [8].
| Table 1: Genetic characteristic of Mucopolysaccharidosis III subtypes. | ||||
| Disease name | affected Gene | Chromosome location | OMIM # | Enzyme/protein deficiency |
| Mucopolysaccharidosis type IIIAa | SGSH | 17q25.3 | 252900 | Heparan-N-sulfatase |
| Mucopolysaccharidosis type IIIBa | NAGL | 17q21.2 | 252920 | alpha-N-acetylglucosaminidase |
| Mucopolysaccharidosis type IIICa | HGSNAT | 8p11.2-p11.1 | 252930 | acetyl CoA alpha-glucosaminide acetyltransferase |
| Mucopolysaccharidosis type IIIDa | GNS | 12q14.3 | 252940 | N-acetylglucosamine 6-sulfatase |
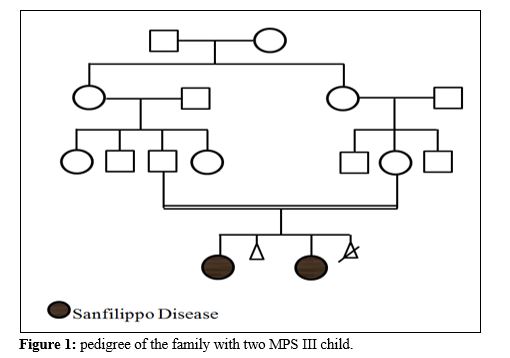
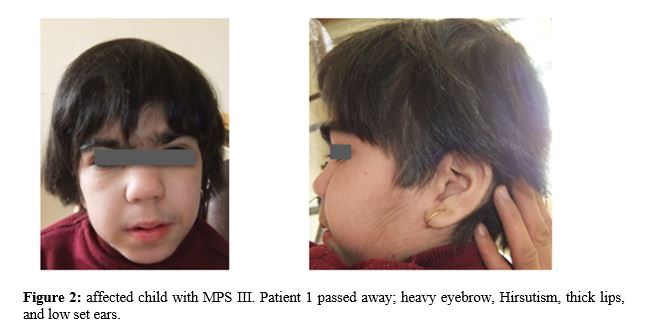
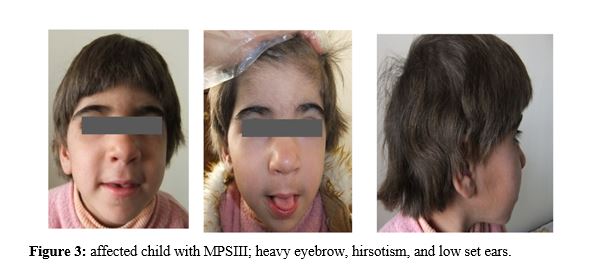
Case presentation
A consanguineous couple with two affected child and one abortion has been refer to welfare genetic counseling center of west Azerbaijan and they will to have a healthy child. First child was 10 years old with profound intellectual disability, history of developmental delay and seizure, low set ears, large tongue, speech deficiency, autistic behavior, facial dysmorphology, short forehead, thick lips, Hirsutism, low hair line, stiff hair and grasping. Second affected child was 7 years old and clinical examination showed development delay, intellectual disability, hyperactivity, hirsutism, large tongue, speech deficiency, short forehead, thick lips and autistic behavior. Clinical examination of both affected children suggested the Lysosomal storage disorders so different diagnosis techniques explained to the family and enzyme activity assay requested. Unfortunately, one of affected child passed away during investigation genetic and genetic analysis carried on just for one affected individual.
| Table 2: Results of functional genetic analysis for MPSIII subtype. | ||||
| Enzyme | Results | Control range | Patient range | unit |
| alpha-N-acetylglucosaminidase | 2.1 | 50-217 | 1.2-6.5 | nmol/17h/mg |
| GlcNAc-6-s sulfatase | 117 | 36-150 | 0-0.7 | nmol/17h/mg |
| Heparan sulphamidase | 60 | 20-90 | 0-1 | nmol/17h/mg |
| AcetylCoA:glucosaminide N-acetyltransferase | 137 | 50-225 | 0.7-4 | nmol/17h/mg |
| β-hexosaminidase(A+B) | 6400 | 4000-23900 | 60-612 | nmol/h/mg |
| β-galactosidase | 629 | 320-1900 | 0-42 | nmol/h/mg |
As family wanted to have a healthy child, all the option for prenatal diagnosis explained and beneficial and damages for each method has been explain, the couple decided to use enzyme activity assay for their fetus so during the 10 week of pregnancy in amniotic fluid has been sent to enzyme activity assay and unfortunately fetus was affected and family ought to abort the baby.
Discussion
Mucopolysaccharidosis and all Lysosomal Storage Disorders present a diagnostic challenge to all healthcare providers. More than 50% of patient with MPS would be recognized by clinical examination but therapeutic intervention and prenatal diagnosis require determining exact involving gene and these challenges can often take several years. Early precise molecular diagnosis can be great significance to the patients and the family. Laboratory study will be needed after clinical diagnosis but genetic analysis has not been the primary tool used in the diagnosis of MPS due to the cost and time requirements [9]. Enzyme activity is one of the most used techniques to diagnosis the MPS III. Reporting family in this paper had two affected child and examined fetus was affected too. Molecular genetics study of patient and the fetus was possible in this family but according the cast and time, family decided to use the enzyme activity assay and the result was satisfied.
Recently Next-Generation Sequencing (NGS) panels have been recommended to the families with various monogenic diseases but NGS study of LSDs has been faced some difficulties [9,10]. Since the technology and applications rapidly change, the available NGS platforms are not yet stable enough [9]. Enzyme activity assay still could be the suitable assay for the diagnosis of LSDs diseases [6]. Genetic counseling for the couple with affected child would be very challenging as there are so many types for MPS and so many options to gather desire consequences. Enzyme detection, molecular genetics and NGS study all would detect the mutated genes but the price and the time management make some limitation for the couples. Molecular genetics study and enzyme activity assay has almost same price but NGS in our county is very expensive [9,10] although it is going to be less over time [6] so, depend on couple financial situation and the time limitation genetic counseling would have been changed to achieve desire results [11].
Acknowledgements
The authors extend their gratitude to the patient and their willingness to participate in this study.
Funding
This article is result of PhD thesis project that has been founded by Kawsar human genetic research center (KHGRC), Tehran, Iran.
Conflict of Interest
The authors hereby affirm that the manuscript is original, that all statements asserted as facts are based on authors careful investigation and accuracy, that the manuscript has not been published and has not been submitted or considered for publication elsewhere, and that authors have full power authority to enter into this copyright assignment and to make the grants herein content.
Ethical approval
All used procedures were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration in 2000. Informed consent was obtained from all patients for being included in the study.
Informed consent
Informed consent was obtained from the reporting patient guardian for being included in the study.
References
- Ferreira CR, Gahl WA. 2016. Lysosomal storage diseases. Translational Science of Rare Diseases. 1-71. [Ref.]
- Bobillo Lobato J, Jiménez Hidalgo M, Jiménez Jiménez L. 2016. Biomarkers in Lysosomal Storage Diseases. Diseases. 4: 40. [Ref.]
- Muenzer J. 2011. Overview of the mucopolysaccharidoses. Rheumatology (Oxford). 5: 4-12. [Ref.]
- White K, Kim T, Neufeld JA. 2010. Clinical assessment and treatment of carpal tunnel syndrome in the mucopolysaccharidoses. J Pediatr Rehabil Med. 3: 57-62. [Ref.]
- Xu M, Motabar O, Ferrer M, et al. 2016. Disease models for the development of therapies for lysosomal storage diseases. Annals of the New York Academy of Sciences. 1371:15-29.[Ref.]
- Fernandez-Marmiesse A, Morey M, Pineda M, et al. 2014. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet journal of rare diseases. 9: 59. [Ref.]
- Yu C, Sun Q, Zhou H. 2016. Enzymatic Screening and Diagnosis of Lysosomal Storage Diseases. North American journal of medicine & science. 6:186-193. [Ref.]
- Di Fruscio G, Schulz A, De Cegli R, et al. 2015. Lysoplex: An efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy. 11: 928-938. [Ref.]
- Komlosi K, Sólyom A, Beck M. 2016. The Role of Next-Generation Sequencing in the Diagnosis of Lysosomal Storage Disorders. Journal of Inborn Errors of Metabolism and Screening. [Ref.]
- Yubero D, Brandi N, Ormazabal A, et al. 2016. Targeted Next Generation Sequencing in Patients with Inborn Errors of Metabolism. PloS one. [Ref.]
- Stewart FJ, Bentley A, Burton BK, et al. 2016. Pregnancy in patients with mucopolysaccharidosis: a case series. Molecular genetics and metabolism reports. 8: 111-115. [Ref.]


















