
-
Mail us:
editor@raftpubs.org
Indexing & Abstracting
Full Text
Research ArticleDOI Number : 10.36811/ijbm.2019.110010Article Views : 7Article Downloads : 12
X-chromosome inactivation: dosage compensation of genes or heterochromatin?
AI Ibraimov
Laboratory of Human Genetics, National Center of Cardiology and Internal Medicine, Bishkek, Kyrgyzstan
*Corresponding author: A.I. Ibraimov, Laboratory of Human Genetics, National Center of Cardiology and Internal Medicine, Bishkek, Kyrgyzstan. Email: ibraimov_abyt@mail.ru
Article Information
Aritcle Type: Research Article
Citation: AI Ibraimov. 2019. X-chromosome inactivation: dosage compensation of genes or heterochromatin?. Int J Biol Med. 1: 75-87.
Copyright:This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Copyright © 2019; AI Ibraimov
Publication history:
Received date: 21 May, 2019Accepted date: 04 June, 2019
Published date: 06 June, 2019
Abstract
X-chromosome inactivation (XCI) is the process by which one of two X chromosomes in mammalian female cells is inactivated. The DNA of the inactive X chromosome is packaged in transcriptionally inactive heterochromatin. It is generally accepted that XCI have evolved to enable dosage compensation in mammals as a way to equalize X-linked gene expression between XX and XY individuals. However, there remain several controversial issues regarding the causes of XCI. The most important of them, why dosage compensation of genes? An alternative hypothesis is discussed that XCI is caused by dose compensation for heterochromatin, rather than genes, in the genome of female mammals due to the lack of a sex chromosome in their karyotype with a large constitutive heterochromatin block, as in Y chromosome in males. It is for this reason that heterochromatinization of the euchromatin regions of one of the X chromosomes occurs. The biological meaning of heterochromatinization is to increase the density of condensed chromatin (??) around the interphase nucleus, responsible for removing excess heat from the nucleus into the cytoplasm, since the compaction of ?? depends on the amount of heterochromatin. The consequence of this process is the inactivation of genes that were in the area of heterochromatinization of the X chromosome.
Keywords: X-chromosome inactivation; Lyonization; Gene dosage compensation; Heterochromatin dosage compensation; Cell thermoregulation
Introduction
X-chromosome inactivation (lyonization) is a process in which one of two X chromosomes in mammalian female cells is inactivated. The DNA of the inactive X chromosome is packaged in transcriptionally inactive heterochromatin. It is believed that X-chromosome inactivation (XCI) have evolved to enable dosage compensation in marsupial and placental mammals as a way to equalize X-linked gene expression between XX and XY individuals. XCI mechanisms have been the subject of intensive research for more than half a century and significant progress has been made in this direction [1].
However, several questions remain regarding the causes of XCI. The most important of them, why dosage compensation of genes? We, in particular, believe that with XCI, not dosage compensation of genes occurs, but ? dosage compensation of the amount of heterochromatin in mammalian genomes. There are other questions, some of which are listed below.
Facts that raise doubts about the causes of XCI in mammals
1) Among the higher eukaryotes, including homeotherm animals, XCI occurs for some reason only in mammals.
2) Chromosome inactivation does not occur in autosomes even in cases where there is a clear excess dose of genes in the genome. For example, in trisomy of autosomes in humans there is no inactivation of the extra chromosome.
3) XCI does not occur in the germ cells of females, where in all oocytes both X-chromosomes are active.
4) It is known, that the mammalian X chromosome is very large, harboring >1,000 genes in mice and humans. It is believed that a double dose of some of these genes is clearly problematic because failure to induce XCI in XX embryos leads to early lethality during development [2, 3, 4]. If all genes are not inactivated on a heterochromatinized X chromosome, then why do mammals need XCI at all?
5) The XCI also displays some degree of epigenetic plasticity in pathological contexts such as cancer. For example, in tumors, the Barr body appears to be absent [5].
Facts supporting views on a dosage compensation of heterochromatin
It is known, that in somatic nuclei of female mammals, the two X chromosomes display very different chromatin states: one X is typically euchromatic and transcriptionally active, and the other is mostly silent and forms a cytologically detectable heterochromatic structure (Barr body).
We believe that the biological meaning of XCI is to compensate for the dose of heterochromatin in the genome of female mammals, in which the total amount of constitutive heterochromatin is usually less than that of males. The following facts speak in favor of such an assumption.
1) The sex chromosomes of mammals are carriers of the largest blocks of chromosomal heterochromatin regions (HRs).
2) The amount of heterochromatin in the genome of men was found to be significantly higher than in women due to the largest block of constitutive heterochromatin on the Y chromosome [6].
3) There are two groups of data indicating the existence of dose compensation for chromosomal heterochromatin regions (HRs) in the human genome. The first group relates to the data on the distribution of the amount of chromosomal Q-heterochromatin regions (Q-HRs) on autosomes in women [7]. The second group of data relates to the relationship between the Y chromosome size and the amount of autosomal Q-HRs [8]. These data are obtained on human populations living permanently in different climatogeographic conditions of Eurasia and Africa. Unfortunately, such studies cannot be carried out on chromosomal C-heterochromatin regions (C-HRs) due to the fact that after C-staining, most chromosomes in the human karyotype are not identified.
The first group of data shows that at the population level the total number of chromosomal Q-HRs on autosomes of women is significantly higher than Q-HRs on autosomes in men. Currently the following quantitative characteristics of chromosomal Q-heterochromatin polymorphism is used in comparative population studies:
a) the distribution of the numbers of Q-HRs in a population, i.e., distribution of individuals having different numbers of Q-HRs in the karyotype regardless of the location (distribution of Q-HRs), which also reflected the range of Q-HRs variability in the population genome;
b) the mean number of chromosomal Q-HRs per individual, as determined by dividing the total number of Q-HRs detected in a given sample by the number of individuals studied;
c) the distribution of Q-HRs on autosomes according to their size and intensity of fluorescence (type of Q-HRs), estimated as described by the Paris Conference (1971, 1975) [6];
d) the size of the Y chromosome, being (a) large (Y=F), (b) medium (F>Y>G), and (c) small (Y=G).
Table 1 shows the distribution of the numbers and mean number of chromosomal Q-HRs on autosomes in males and females in two age groups of Kazakh nationality.
| Table 1: Distribution of the numbers and mean number of Q-HRs on autosomes in males and females in newborns and 18 - 25 years individuals. | ||||
|---|---|---|---|---|
| Number of Q-HRs | Newborns Boys (n=207) I | Newborns Girls(n=182)II | Males 18 - 25 years (n=49)III | Females 18 - 25 years(n=190)IV |
| 0 | 3 | 1 | ||
| 1 | 5 | 4 | 9 | 7 |
| 2 | 39 | 21 | 12 | 24 |
| 3 | 47 | 38 | 13 | 45 |
| 4 | 51 | 46 | 11 | 49 |
| 5 | 37 | 39 | 4 | 40 |
| 6 | 18 | 20 | 17 | |
| 7 | 7 | 13 | 7 | |
| Total number of Q-HRs | 770 | 750 | 136 | 745 |
| Mean number of Q-HRs | 3.72 ± 0.102 | 4.12 ± 0.111 | 2.78 ± 0.176 | 3.92 ± 0.104 |
| Statistics | 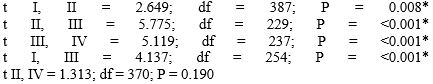 |
|||
| * - these differences are statistically significant. | ||||
As can be seen from this Table, in every case females are characterized by the highest mean number value, and by a broad range of variability in the distribution of the numbers of chromosomal Q-HRs as compared to males. These differences are statistically significant. The same data were obtained for other racial and ethnic groups [7]. The amount of chromosomal Q-HRs in the human genome can be additionally evaluated according to their size and intensity of fluorescence on a five-point degree [6]. Later, recommendations of the Paris Conference on this issue have been further developed in a number of papers [9-12] and comparative population studies began to consider Q-HRs with only 4 and 5 degrees of fluorescence intensity. In estimating the size of Q-HRs we adhered to the recommendations of Yamada and Hasegawa (1978) according to which the sizes of Q-bands were divided into five degrees, comparing them with the short arm of chromosome 18. However, mostly Q-HRs of medium (> 0.5-0.75 × 18p) and large (> 0.75 - 1,0 × 18p) sizes are found in a population. Abbreviation “QFQ” stands for “Q-bands by fluorescence using quinacrine” [6]. For example, “QFQ 15” indicates that the given Q-band has the medium size with very bright fluorescence. Table 2 presents data on the size and intensity of fluorescence of chromosomal Q-HRs in newborns and young adults (18-25-year-olds) of the Kazakh nationality. In the examined samples of individuals QFQ points ranged from 15 to 112. Here QFQ 15 indicates that the genome of a given individual has one Q-HR of medium size with the intensity of fluorescence equal to 5 degrees. While, QFQ 98 means that the karyotype of this individual has seven Q-HRs of medium size with the fluorescence intensity of 5 degrees. For statistical analysis types of Q-HRs we divided into 7 groups: Group 1 (14, 15 points); Group 2 (28, 29 points), Group 3 (40 to 45 points); Group 4 (53 to 58 points); Group 5 (from 67 to 73 points), Group 6 (80 to 86 points); Group 7 (98 to 112 points). In other words, Group 1 included individuals the karyotype of which had only one Q-HR, Group 2 with two, and the remaining groups with three or more Q-bands with intensity of fluorescence of 5 degrees.
| Table 2: Types of Q-HRs on autosomes in males and females in newborns and 18 - 25 years individuals of Kazakh nationality. | ||||
|---|---|---|---|---|
| Types of Q-HRs | Newborns Boys (n = 180) I | Newborns Boys (n = 159) II | Males 18-25 years (n = 43) III | Females 18-25 years(n = 120)IV |
| 0 | 3 | 2 | ||
| 1 | 6 | 4 | 8 | 7 |
| 2 | 34 | 15 | 7 | 14 |
| 3 | 40 | 33 | 13 | 28 |
| 4 | 42 | 36 | 12 | 30 |
| 5 | 36 | 40 | 2 | 26 |
| 6 | 14 | 14 | 1 | 11 |
| 7 | 5 | 13 | 4 | |
| 8 | 4 | |||
| Total number of Q-HRs | 661 | 668 | 125 | 463 |
| Mean number of Q-HRs | 3.67 ± 0.110 | 4.20 ± 0.668 | 2.91 ± 0.193 | 3.83 ± 0.132 |
| Statistics | t I, II = 3.164; df = 337; P = 0.002* t III, IV = 3.807; df = 161; P = <0.001* |
|||
| *These differences are statistically significant. | ||||
As shown in this Table, in the genome of females the amount of chromosomal Q-HRs significant more not only in number but also in size and intensity of fluorescence. Similar data were obtained in other samples [7]. The second group of data indicating the existence of heterochromatin dosage compensation is illustrated by the example of the relationship between the Y chromosome size and the number of autosomal Q-HRs in human populations [8]. It should be emphasized that in comparative population cytogenetic studies only autosomal Q-HRs are considered. Nevertheless, variability of the largest Q-heterochromatin band in the human genome, localized on the q12 segment of the Y chromosome, has been mainly considered separately from the quantitative variability of autosomal Q-HRs [9-23]. Here we show the results of our observations on the populations of Eurasia and Africa, which indicate that there is connection between the mean number of autosomal Q-HRs and sizes of Q-heterochromatin blocks on Y chromosome [19,22]. Table 3 presents the distribution of the numbers and mean number of autosomal Q-HRs in males with Y chromosomes of various sizes in Kazakh newborns. The same patterns were found in other racial and ethnic groups [7,8].
| Table 3: Distribution of the numbers and mean number of autosomal Q-HRs in males with Y chromosomes of various sizes Kazakh newborns. | |||
|---|---|---|---|
| Number of Q-HRs | Large Y ≥ F (n = 53)I | Medium F > Y > G (n = 102)II | Small Y ≤ G (n = 32)III |
| 0 | 3 | ||
| 1 | 5 | 1 | |
| 2 | 21 | 12 | 2 |
| 3 | 11 | 26 | 1 |
| 4 | 6 | 28 | 10 |
| 5 | 7 | 22 | 13 |
| 6 | 10 | 4 | |
| 7 | 3 | 2 | |
| Total number of Q-HRs | 139 | 406 | 150 |
| Mean number of Q-HRs | 2.62 ± 0.185 | 3.98 ± 0.129 | 4.69 ± 0.203 |
| Statistics | t I, II = 6.077; df = 153; P = <0.001* t II,III = 2.748; df = 132; P = 0.007* t I,III = 7.223; df = 83; P = <0.001* |
||
| * these differences are statistically significant. | |||
The existence of a close connection between the number of Q-HRs on autosomes and the size of Q-heterochromatin on the Y chromosome were showed on adult individuals, representing all three racial and ethnic groups living permanently in Eurasia and Africa [22]. Table 4 shows the distribution of the numbers and the mean number of Q-HRs on autosomes in males having different Y chromosome sizes. Significant decreases in mean numbers in males with large Y chromosomes were noted. Of interest is the fact that in the group of males with large Y chromosomes narrowing of the distribution of numbers of autosomal Q-HRs was evident, as well as its ‘upwards’ shift during the greatest range of variability in the number of Q-HRs in the karyotype in groups with medium and small Y chromosomes. Males with large Y chromosomes were characterized by low values in the mean number of Q-HRs per individual and by a low range of variability in the distribution of Q-HRs compared with males with medium and especially small Y chromosomes. These differences are statistically significant.
| Table : Distribution of the numbers and mean number of autosomal Q-HRs in males with Y chromosomes of various sizes (Ibraimov et al., 2000). | ||||
|---|---|---|---|---|
| Populations | Number of Q-HR | Y ≥ F (n = 30) I |
F > Y > G (n = 261) II |
Y ≤ G (n = 36) III |
| Negroes of Africa (Mozambique, Guinea-Bissau, Zimbabwe, Angola) | 0 | |||
| 1 | 1 | |||
| 2 | 5 | 16 | ||
| 3 | 5 | 42 | 7 | |
| 4 | 7 | 64 | 7 | |
| 5 | 7 | 66 | 6 | |
| 6 | 1 | 46 | 8 | |
| 7 | 4 | 20 | 5 | |
| 8 | 4 | 2 | ||
| 9 | 2 | 1 | ||
| 10 | 1 | |||
| Total number of Q-HRs | 123 | 1 220 | 187 | |
| Mean number of Q-HRs | 4.10 ± 0.30 | 4.67 ± 0.09 | 5.10 ± 0.28 | |
| t I, II = 1.98; df = 289; P = <0.049 Statistics t II, III = 1.61; df = 295; P = <0.108; t I,III = 2.66; df = 64; P = <0.009; |
||||
| Continued | ||||
| Populations | Number of Q-HRs | Y ≥ F (n = 20) I |
F > Y > G (n = 78) II |
Y ≤ G (n = 16) III |
| Steppe Mongoloids (Kazakhs, Chinese) | 0 | 2 | ||
| 1 | 7 | 11 | 2 | |
| 2 | 4 | 26 | 4 | |
| 3 | 4 | 21 | 2 | |
| 4 | 2 | 11 | 3 | |
| 5 | 1 | 4 | 4 | |
| 6 | 5 | 1 | ||
| Total number of Q-HRs | 40 | 220 | 54 | |
| Mean number of Q-HRs | 2.00 ± 0.31 | 2.82 ± 0.15 | 3.38 ± 0.40 | |
| t I,II = 2.39; df = 30; P = <0.021; Statistics t II,III = 1.32; df = 20; P = <0.194; t I,III = 2.75; df = 32; P = <0.008; |
||||
| Continued | ||||
| Populations | Number of Q-HRs | Y ≥ F (n = 56) I |
F > Y > G (n = 280) II |
Y ≤ G (n = 24) III |
| Russians | 0 | 10 | 23 | 1 |
| 1 | 21 | 53 | 5 | |
| 2 | 14 | 103 | 6 | |
| 3 | 7 | 65 | 7 | |
| 4 | 4 | 25 | 2 | |
| 5 | 9 | 2 | ||
| 6 | 2 | 1 | ||
| Total number of Q-HRs | 86 | 611 | 62 | |
| Mean number of Q-HRs | 1.53 ± 0.15 | 2.18 ± 0.07 | 2.58 ± 0.29 | |
| Statistics | t I,II = 3.93; df = 82; P = <0.000; t II,III = 1.34; df = 26; P = <0.183; t I,III = 3.14; df = 37; P = <0.003; |
|||
| Continued | ||||
| Populations | Number of Q-HRs | Y ≥ F (n = 16) I |
F > Y > G (n = 125) II |
Y ≤ G (n = 11) III |
| Kyrgyz of Pamir and Tien-Shan | 0 | 3 | 11 | |
| 1 | 5 | 26 | 3 | |
| 2 | 6 | 49 | 4 | |
| 3 | 2 | 20 | 3 | |
| 4 | 14 | 1 | ||
| 5 | 3 | |||
| 6 | 2 | |||
| Total number of Q-HRs | 23 | 267 | 24 | |
| Mean number of Q-HRs | 1.43 ± 0.24 | 2.13 ± 0.11 | 2.18 ± 0.29 | |
| t I, II = 2.12; df = 139; P = <0.036; Statistics t II,III = 0.13; df = 134; P = <0.900; t I,III = 1.97; df = 25; P = <0.060; |
||||
| Continued | ||||
| Populations | Number of Q-HRs | Y ≥ F (n = 56) I |
F > Y > G (n = 215) II |
Y ≤ G (n = 26) III |
| Northern Mongoloids (Chukchi, Yakuts, Khakass, Nenets, Selkups) | 0 | 10 | 24 | |
| 1 | 22 | 50 | 6 | |
| 2 | 12 | 71 | 9 | |
| 3 | 6 | 46 | 9 | |
| 4 | 6 | 17 | 2 | |
| 5 | 7 | |||
| Total number of Q-HRs | 88 | 433 | 59 | |
| Mean number of Q-HRs | 1.57 ± 0.16 | 2.01 ± 0.08 | 2.27 ± 0.18 | |
| t I,II = 2.48; df = 269; P = <0.017; Statistics t II,III = 1.29; df = 38; P = <0.198; t I,III = 2.88; df = 66; P = <0.005; |
||||
It is generally known that the size of the Q-heterochromatin on the long arm of a Y chromosome of even medium size is greater than that of the Q-HRs on any of seven Q-polymorphic autosomes in the human karyotype, especially as the morphological variability of the Y chromosome (large, medium, small) is mainly determined by the size of the Q-heterochromatin segment on its long arm. Based on the data presented above, we assume, that the Q-heterochromatin block on the Y chromosome, being the largest Q-heterochromatin segment in the human genome, somehow ‘restricts’ the overall amount of Q-HRs on autosomes in males. Apparently, for the same reason, amount of autosomal Q-HRs increases in females compared to males within each individual population. In addition, human chromosomal Q-HRs may have a basically similar role regardless of their location in the karyotype [7,22]. We believe the correct to explain the increasing number of chromosomal Q-HRs on autosomes in females at the population level by the existence of some evolutionary established mechanism that “compensates” the difference in the “dose” of Q-heterochromatin material in the female genome due to the lack of chromosomes in their karyotype, which carries largest Q-HR, as Y chromosome. Apparently, there is some mechanism that limits the “dose” of chromosomal Q-HRs in the human genome to a certain level. Indeed, the human karyotype has 25 loci (3 cen, 4 cen, 13 p11, 13 p13, 14 p11, 14 p13, 15 p11, 15 p13, 21 p11, 21 p13, 22 p11, 22, p13 and Yq12), where Q-heterochromatin can potentially be detected. However, as yet no one could found 25 chromosomal Q-HRs in the human karyotype; usually their number varies from 0 to 10 [12,14,24,25]. These data gave us reason to believe that: 1) Q-heterochromatin on the Y chromosome, being the largest in the human genome, somehow “restricts” the total content of Q-HRs on the autosomes in males; and 2) Q-HRs on human chromosomes appears to have a common nature, regardless of their localization in the karyotype.
Discussion
As it is known, Lyon (1961) [25] proposed the single-active X-chromosome hypothesis to explain the observation that in the mouse, females heterozygous for X-linked fur color genes are patchy mosaics of two colors. To quote Lyon: “... (1) that the heteropicnotic X-chromosome can be either paternal or maternal in origin in different cells of the same animal; (2) that it is genetically inactivated”. According to Lyon this mechanism provides dosage compensation for X-linked genes because each cell, male or female, has only one X-chromosome that is transcribed. We believe that the Lyon’s hypothesis, although flawless in terms of logic, nevertheless, it does not fully reflect the essence of XCI. If only the problem was that XCI have evolved to enable dosage compensation in mammals as a way to equalize X-linked gene expression between XX and XY individuals, then all X-linked genes would be inactivated. The point that we are trying to convey is that: a) X-inactivation is not involved in the sex determination, as Lyon stated (1992) [26]; b) ?-chromosome is not being inactivated, but it is heterochromatinized in order to compensate the lacking in the female karyotype the largest block of the constitutive HRs in the interest of the cell thermoregulation (CT).
By CT we mean the elimination of the temperature difference between the nucleus and cytoplasm, when, for one reason or another, the level of thermal energy in the nucleus becomes higher than of the cytoplasm. Based on investigations of condensed chromatin (CC), interphase nucleus and redundant ncDNAs in the genome, an attempt is made to justify the view of possible participation of chromosomal HRs in cell thermoregulation. CC, being the densest domains in a cell, apparently conducts heat between the cytoplasm and nucleus when there is a difference in temperature between them [27,28]. The CT hypothesis has experimental evidence at the level of the human body. In particular, it is shown that individuals in population differ from each other in body heat conductivity (BHC) and its level depends on the amount of chromosomal Q-HRs in human genome (Ibraimov et al., 2014) [7]. As a whole our results show that: a) individuals in a population differ from each other on the level of BHC; b) on the average BHC of males is statistically significantly higher than that of females; c) individuals differ in BHC from different age groups, on the average human BHC level is steadily changed decreasing with age; d) natives of low altitude regions of southern latitude differ on the average by higher BHC than population of high mountains and northern latitude [29-34].
Thus, it would be more correct to speak about compensation of the heterochromatin dosage and not about the dosage (double) of genes. That CT may be related to inactivation of one X-chromosome in humans is evidenced by such fact as the statistically significantly low level of the BHC in women compared to men [31, 32]. This is probably due to the fact that the condensed chromatin (CC) in interphase cells of women does not have the same density as in men. Apparently, facultative heterochromatin of the inactivated X-chromosome is still inferior to constitutive heterochromatin on the Y chromosome on ability to compactize CC in the cells of the female body. The question of the reality of the existence of heterochromatin dose compensation, on the example of human chromosomal Q-HRs, we believe proven. In this case, can we assume that XCI and Q-HRs dose compensation in the genome of women are of the same nature? Our answer: Yes, rather than no.
There are few arguments in favor of such a point of view. However, some of them deserve attention.
1) From a morphological point of view, constitutive and facultative heterochromatin is not significantly different.
2) Once inactivated, the X chromosome is consistently found in close association with the nuclear membrane [35-37]and/or at the periphery of the nucleolus [38,39].
3) Inactivated X chromosome is often found in the nucleolus [38], where chromosomal HRs of autosomes 1, 9, and 16 accumulate, as well as Y chromosome [39], which we consider as components of the CT that are involved in the removal of excess heat from the cell nucleus [40,28].
4) In embryogenesis, XCI begins at the blastocyst stage, that is, at the stage of multicellularity, when the problem of removing excess heat from the nucleus begins [27,41-45]
If we recognize that the tasks of both types of heterochromatin in the cell are the same, then there is no reason to consider their possible biological role separately. In this case, both types of heterochromatin should be involved in CT because their dose is important, not localization in the karyotype.
Concluding remarks
Returning to the beginning of the article, we will try to answer the questions posed there.
1) To the question why among homeotherm animals, XCI occurs only in mammals, can be answered like this; mammalian homeotherms have the highest level of metabolic rate and they are able to maintain fluctuations in body temperature within very narrow limits.
2) Chromosome inactivation does not occur in autosomes even when there is a clear excess dose of genes in the genome (as in trisomy) just because there is no need to compensate for such a big block of constitutive heterochromatin as is available on Y chromosome.
3) XCI does not occur in the germ cells of females, where both X-chromosomes are active in all oocytes because there are high level metabolism rate and active cell proliferation associated with the production of gametes that need the full set of X-linked genes.
4) The answer to the question of why a complete inactivation of all (> 1,000) genes does not occur on an inactive X chromosome can be very simple; the essence of heterochromatinization of the X chromosome is not in gene inactivation, but in compensating for the dose of the missing constitutive heterochromatin in the female genome.
5) Why does the XCI also display some degree of epigenetic plasticity in pathological contexts such as cancer? For example, in tumors, the Barr body appears to be absent. The answer is obvious: in tumors the level of cellular metabolism is very high and rapidly growing tumor cells need many, including all X-linked genes of a given organism.
Thus, the cause of XCI is a dose compensation of heterochromatin, rather than genes, in the genome of female mammals due to the lack in their karyotype of the sex chromosome with a large block of constitutive heterochromatin, as Y chromosome in males. It is for this reason that heterochromatinization of the euchromatin regions of one of the X chromosomes occurs. The biological meaning of heterochromatinization is to increase the density of condensed chromatin around the interphase nucleus (responsible for removing excess heat from the nucleus into the cytoplasm at CT) to compensate for the missing dose of constitutive heterochromatin in the genome of mammalian females, since the compaction of condensed chromatin depends on the amount of chromosomal HRs. The consequence of this process is the inactivation of genes that were in the area of heterochromatinization of the X chromosome.
References
- Galupa R, Heard E. 2018. X-chromosome inactivation: a crossroads between chromosome architecture and gene regulation. Annu Rev Genet. 52: 535-66. [Ref.]
- Takagi N, Sugawara O, Sasaki M. 1982. Regional and temporal changes in the pattern of X-chromosome replication during the early post-implantation development of the female mouse. Chromosoma. 85: 275-286. [Ref.]
- Tada T, Takagi N, Adler ID. 1993. Parental imprinting on the mouse X chromosome: effects on the early development of X0, XXY and XXX embryos. Genet Res. 62: 139-48.[Ref.]
- Marahrens Y, Panning B, Dausman J, et al. 1997. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 11:156-66. [Ref.]
- Chaligne R, Heard E. 2014. X-chromosome inactivation in development and cancer. FEBS Lett. 588: 2514-2522. [Ref.]
- Paris Conference. (1971) and Supplement (1975). Standartization in human cytogenetics. Birth Defects: Original Article Series. XI. 1-84. The National Foundation. New York. [Ref.]
- Ibraimov AI, Akanov AA, Meimanaliev TS, et al. 2014. Human Chromosomal Q-heterochromatin Polymorphism and Its Relation to Body Heat Conductivity. Int J Genet. 6: 142-148. [Ref.]
- Ibraimov ?I, Karagulova G?, Kim ?Y. 2000. The relationship between the Y chromosome size and the amount of autosomal Q-heterochromatin in human populations. Cytobios. 102: 35-53. [Ref.]
- Müller ? J, Klinger ?P, Glasser ?. 1975. Chromosome polymorphism in ? human newborn population. II. Potentials of polymorphic chromosome variants for characterizing the idiograms of an individual. Cytogenet Cell Genet. 15:239-255. [Ref.]
- Buckton KE, O’Riordan ML, Jacobs PA, et al. 1976. C- and Q-band polymorphisms in the chromosomes of three human populations. Ann Hum Genet 40: 90-112. [Ref.]
- Lubs HA, Patil SR, Kimberling WJ, et al. 1977. Racial differences in the frequency of Q- and C-chromosomal heteromorphism. Nature. 268: 631-632. [Ref.]
- Yamada ?, Hasegawa ?. 1978. Types and frequencies of Q-variant chromosomes in ? Japanese population. ?um Genet. 44: 89-98. [Ref.]
- Geraedts JP?, Pearson PL. 1974. Fluorescent chromosome polymorphism: frequencies and segregation in ? Dutch population. Clin Genet 6: 247-257. [Ref.]
- Al-Nassar ??, Palmer ?G, Connealy P?, et al. 1981. The genetic structure of the Kuwaiti population. II. The distribution of Q-band chromosomal heteromorphisms. ?um Genet 57: 423-427. [Ref.]
- Ibraimov ?I, Mirrakhimov ??. 1982a. Human chromosomal polymorphism. III. Chromosomal Q-polymorphism in Mongoloids of Northern Asia. Hum Genet. 62: 252-257. [Ref.]
- Ibraimov ?I, Mirrakhimov ??. 1982b. Human chromosomal polymorphism. IV.Q-polymorphism in Russians living in Kirghizia. Hum Genet. 62: 258-260. [Ref.]
- Ibraimov ?I, Mirrakhimov ??. 1982c. Human chromosomal polymorphism. V. Chromosomal Q-polymorphism in African populations. Hum Genet. 62: 261-265. [Ref.]
- Ibraimov ?I, Mirrakhimov ??, Nazarenko S?, et al. 1982. ?uman chromosomal polymorphism. I. Chromosomal Q-polymorphism in Mongoloid populations of Central Asia. Hum Genet. 60: 1-7. [Ref.]
- Ibraimov ?I, Mirrakhimov ??, Axenrod ?I, et al. 1986. Human chromosomal polymorphism. IX. Further data on the possible selective value of chromosomal Q-heterochromatin material. Hum Genet. 73: 151-156. [Ref.]
- Ibraimov ?I, Kurmanova GU, Ginsburg ??, et al. 1990. Chromosomal Q-heterochromatin regions in native highlanders of Pamir and Tien-Shan and in newcomers. Cytobios. 63: 71-82. [Ref.]
- Ibraimov ?I, Axenrod ?I, Kurmanova GU, et al. 1991. Chromosomal Q-heterochromatin regions in the indigenous population of the Northern part of West Siberia and in new migrants. Cytobios. 67: 95-100. [Ref.]
- Ibraimov ?I, Karagulova G?, Kim ?Y. 2000. The relationship between the Y chromosome size and the amount of autosomal Q-heterochromatin in human populations. Cytobios. 102: 35-53. [Ref.]
- Ibraimov AI, Akanov AA, Meymanaliev TS, et al. 2013. Chromosomal Q-heterochromatin polymorphisms in 3 ethnic groups (Kazakhs, Russians and Uygurs) of Kazakhstan. Int J Genet. 5:121-124. [Ref.]
- Ibraimov ?I, Mirrakhimov ??. 1985. Q-band polymorphism in the autosomes and the Y chromosome in human populations. In: “Progress and Topics in Cytogenetics. The Y chromosome. Part ?. Basic characteristics of Y chromosome”. ?. ?. Sandberg (Ed). Alan R. Liss, Inc., New York. USA. 213-287. [Ref.]
- Ibraimov AI. 2010. Chromosomal Q-heterochromatin regions in populations and human adaptation. In: MK Bhasin, C Susanne (Eds.): Anthropology Today: Trends and Scope of Human Biology. Delhi: Kamla- Raj Enterprises. 225-250. [Ref.]
- Lyon MF. 1992. Some milestones in the history of X chromosome inactivation. Annu Rev Genet, 26: 17-28. [Ref.]
- Ibraimov AI. 2003. Condensed chromatin and cell thermoregulation. Complexus. 1: 164-170. [Ref.]
- Ibraimov AI. 2017. Cell Thermoregulation: Problems, Advances and Perspectives. J Mol Biol Res. 7: 58-79. [Ref.]
- Ibraimov AI. 2018. Human Body Heat Conductivity in norm and pathology: A review. Advance Research Journal of Multidisciplinary Discoveries. 32: 12-21. [Ref.]
- Ibraimov ?I. 2019. Human adaptation: why only genes? Int J Biol Med. 1: 22-33. [Ref.]
- Ibraimov AI, Tabaldiev SK. 2007. Condensed chromatin, cell thermoregulation and human body heat conductivity. J Hum Ecol. 21: 1-22. [Ref.]
- Ibraimov AI, AK Kazakova, IK Moldotashev, et al. 2010a. Variability of Human Body Heat Conductivity in Population. I. Methodological and Theoretical Approaches. J. Hum. Ecol. 32: 1-22. [Ref.]
- Ibraimov AI, AK Kazakova, IK Moldotashev, et al. 2010b. Variability of Human Body Heat Conductivity in Population. II. Diseases of Civilization. J. Hum. Ecol. 32: 69-78. [Ref.]
- Ibraimov AI. 2014. Chromosomal Q-heterochromatin and sex in human population. J. Mol. Biol. Res. 4: 10-19. [Ref.]
- Klinger HP. 1958. The fine structure of the sex chromatin body. Exp Cell Res 14: 207-211. [Ref.]
- Hoehn H, Martin GM. 1973. Nonrandom arrangement of human chromatin: topography of disomic markers X, Y, and 1h+. Cytogenet Genome Res. 12: 443-452. [Ref.]
- Belmont AS, Bignone F, Ts’o POP. 1986. The relative intranuclear positions of Barr bodies in XXX non-transformed human fibroblasts. Exp Cell Res. 16:165-79. [Ref.]
- Bourgeois CA, Laquerriere F, Hemon D, et al. 1985. New data on the in-situ position of the inactive X chromosome in the interphase nucleus of human fibroblasts. Hum Genet. 69:122-29. [Ref.]
- Schmid, M., Vogel, W., Krone, W. 1975. Attraction between centric heterochromatin of human chromosomes. Cytogenet Cell Genet, 15: 66-80. [Ref.]
- Zhang L-F, Huynh KD, Lee JT. 2007. Perinucleolar targeting of the inactive X during S phase: evidence for a role in the maintenance of silencing. Cell. 129: 693-706. [Ref.]
- Ibraimov AI. 2015. Heterochromatin: The visible with many invisible effects. Global Journal of Medical Research. 15: 7-32. [Ref.]
- Ibraimov AI. 2004. The origin of condensed chromatin, cell thermoregulation and multicellularity. Complexus. 2: 23-34. [Ref.]
- Borensztein M, Syx L, Ancelin K, et al. 2017. Xist-dependent imprinted X inactivation and the early developmental consequences of its failure. Nat Struct Mol. Biol. 24: 226-33. [Ref.]
- Ibraimov AI, Karagulova GO, Kim EY. 1997. Chromosomal Q-heterochromatin regions in indigenous populations of the Northern India. Ind J Hum Genet, 3: 77-81.[Ref.]
- Takagi N, Abe K. 1990. Detrimental effects of two active X chromosomes on early mouse development. Development. 109: 189-201. [Ref.]


















